What You Need to Know About CSV in the Life Sciences Industry
On December 15, 2021
Computer System Validation, or “CSV”, is a process of testing and qualifying a computerized system to ensure that it works as intended in a consistent and reproducible manner that is as safe, secure, and reliable.
CSV is a top priority for pharmaceutical, biotech, and medical device companies, which must show that all computer systems can ensure patient safety, product efficacy, and data integrity by being fit for their intended uses.
What Are the Purposes of CSV?
CSV is the FDA’s response to an increasingly growing demand from the Life Sciences industry to use paperless systems. The purpose of CSV is to reduce and mitigate compliance risks before a particular system goes live and maintain compliance when computer systems are modified or upgraded.
What Are the Benefits of CSV in Pharma?
There are many benefits, but here are the most significant ones:
Business Continuity: CSV helps identify system weaknesses before they become major problems and, thus, reduce the downtime
Compliance: CSV demonstrates your commitment to complying with guidelines set by regulatory agencies.
Peace of Mind: By completing CSV, you’re ensuring that your computer software performs consistently and is fit for its intended use.
How Is CSV done?
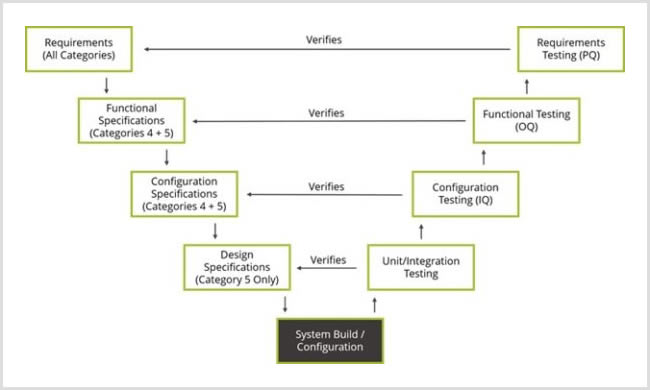
Any Life Sciences company must perform CSV in line with FDA and other global regulatory guidelines. The level of documentation required is typically addressed through adherence to the GAMP-5 V-model:
Figure 1: GAMP 5 V-model, highlighting the documentation that is required based on the applicable category, where Category 1 is tied to Infrastructure software, Category 3 is non-configurable software, Category 4 is configured software, and Category 5 is custom software.
Create a Validation Plan
Think of the validation plan as a strategy. It outlines the approach you’ll take to CSV and what steps you must take to meet your objectives. It should include clear outcomes, acceptance criteria, and required activities.
Define User Requirements Specifications (URS)
The URS defines clearly and precisely what the user wants the system to do and what attributes it should have. Additionally, it describes any non-functional requirements and constraints. It should be driven by the business process needs and include requirements related to patient safety, product quality, and data integrity.
Specify Functional Specifications (FS)
The FS defines the full system functionality, including how the user and business requirements are satisfied. It is the basis for the system design, customization, development, and testing.
Based on the type of system, these specifications, if needed, provide a detailed technical expansion of the FS.
The DS is the activity that involves both hardware and software as a combined document.
The CS details the configuration parameters and how these settings address the requirements in the URS.
Write IQ/OQ/PQ Tests Scripts
Installation Qualification (IQ): The process of validating that the system is successfully installed according to specifications and requirements in the specified environment.
Operational Qualification (OQ): The process of validating that the system is operating as intended and that requirements are verified/tested against the FS document.
Performance Qualification (PQ): The process of validating that the system is performing satisfactorily for its intended purpose and that requirements are verified/tested against the URS document.
Create a Validation Summary Report
The validation summary report summarizes your CSV process. For example, it will describe:
Activities carried out during the Validation
Any deviations made (and the justifications for the changes)
Limitations and restrictions
Incidents
Corrective actions
Maintain System Release Documentation
Even once the system has been accepted and released for use, there are still maintenance needs to consider. Compliance is achieved through using up-to-date documented procedures. All documentation and training should cover use, maintenance, and management in activities such as:
Security and System Administration
Change Management Process
Backup and Restore
Periodic Review
Data Archival and Retrieval
How Can AX for Pharma Support Your CSV Compliance Effort?
AX for Pharma follows a risk-based approach to CSV called Computer Software Assurance (CSA). This streamlined, pragmatic approach meets regulatory requirements by critically assessing relevant risks and confirming that testing, documentation, and evidence collection align with any risks that have been identified.
By choosing AX for Pharma as your partner, you’ll benefit from our compliant framework and commitment to good practices. We also offer a set of tools to simplify the validation process and reduce the effort to achieve compliance.
Contact us to discuss computer system validation in Life Sciences, and how we might help.
Support for CSV in Life Sciences Industries | AX for Pharma 365™
Although the validation process may seem overwhelming, AX for Pharma can help. Our experienced team will give you the guidance you need to complete your CSV and meet your compliance requirements. Whether you’re in Life Sciences, we have a CSV solution for your needs. Contact us today to learn more.
We use cookies to improve your user experience on our website. By browsing our website, you consent to our use of cookies. For more information, please visit our privacy policy.
Thank you.I Agree